 期刊:Science China-Life Sciences
期刊:Science China-Life Sciences
影響因子:8.0
成簇的規則間隔短回文重復序列 (CRISPR)/Cas9 系統是編輯哺乳動物基因組的革命性方法。隨著慢病毒遞送方法的發展,CRISPR 篩選技術出現,并實現了全基因組敲除的低成本方式。然而,CRISPR 篩選只能分析具有非常不同表型的基因,例如那些顯著影響細胞生長的基因,或者那些可以用抗體或熒光蛋白直接檢測到的基因。
2016 年,開發了一種稱為單細胞 CRISPR 篩選 (scCRISPR-seq) 的新技術,該技術將 CRISPR 擾動和單細胞測序相結合,以實現大規模單細胞分辨率的混合遺傳篩選。scCRISPR-seq 的關鍵技術創新是慢病毒載體的創造性設計,稱為 Perturb-seq 載體,允許從測序中鑒定每個細胞中的 sgRNA(圖 16A)。scCRISPR-seq 可以促進復雜調控機制和異質細胞群的高通量功能解剖。
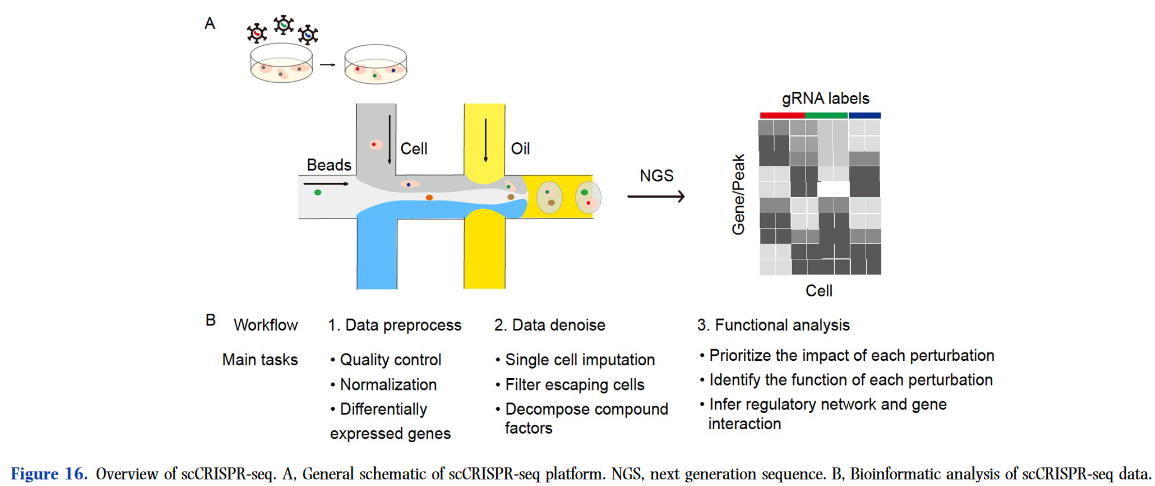
在本章中,我們將分四個不同的部分全面回顧 scCRISPR-seq。首先,我們將介紹 scCRISPR-seq 每個類別中的代表性技術。其次,我們將深入研究專門為分析 scCRISPR-seq 數據開發的主要工具。第三,我們將探索 scCRISPR-seq 的重要應用。最后,我們將得出結論并參與與 scCRISPR-seq 相關的局限性和未來趨勢的討論。
scCRISPR-seq 平臺類別
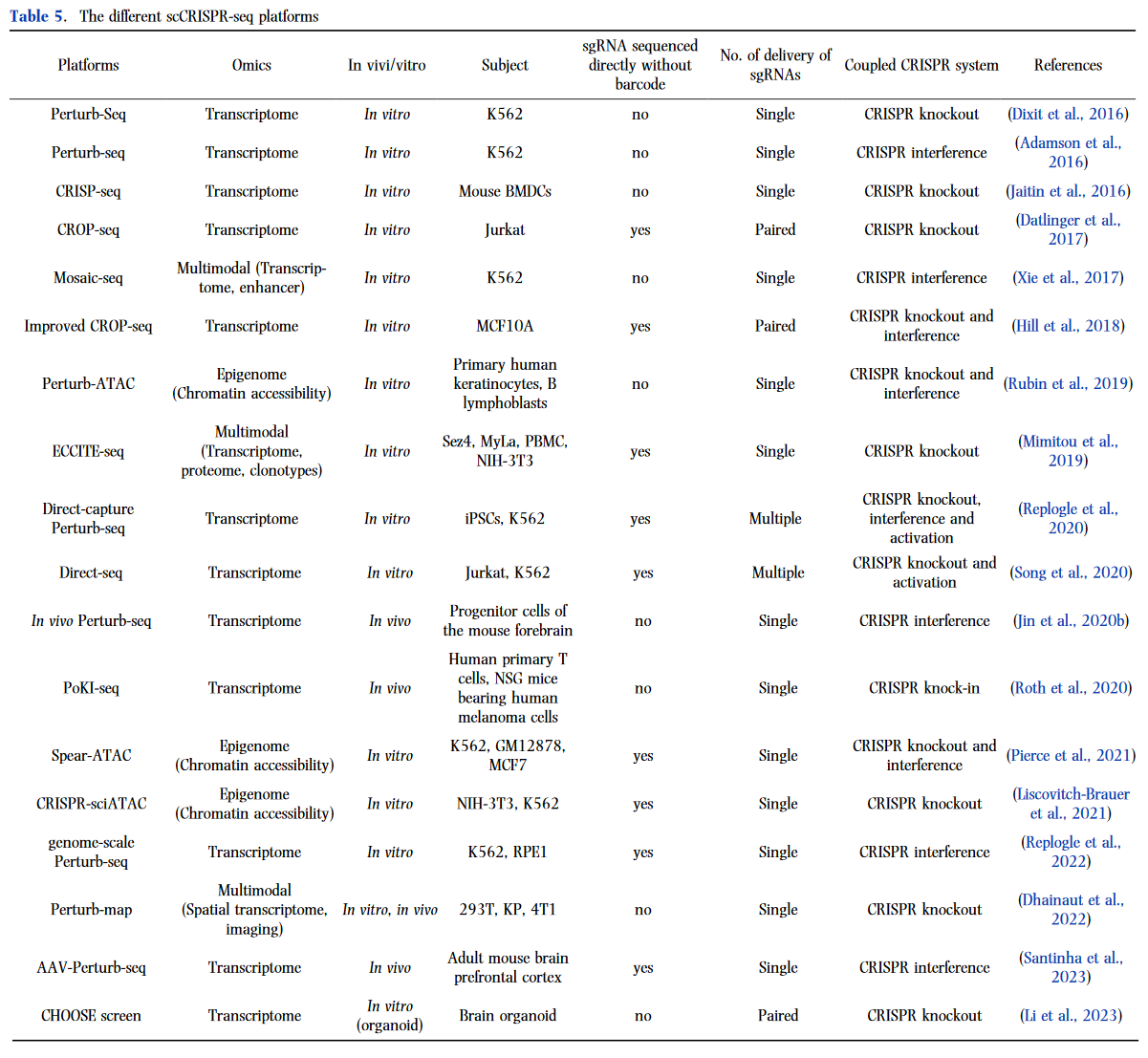
目前,已經出現了許多替代的 scCRISPR-seq 平臺(表 5)。基于 scCRISPR-seq 的集成組學方法,這些平臺可分為三大類:基于轉錄組的 scCRISPR-seq、基于表觀基因組的 scCRISPR-seq 和多模態 scCRISPR-seq。
基于轉錄組的 scCRISPR-seq
主要的 scCRISPR-seq 平臺是基于轉錄組的應用程序,將 CRISPR 篩選與單細胞 RNA-seq 相結合。對于基于轉錄組的 scCRISPR-seq,Perturbseq 載體通常由單向導 RNA (sgRNA)、細胞條形碼 (CBC)、基因條形碼 (GBC) 和 UMI 組成,例如 Perturb-seq和 CRISPseq。在 Perturb-seq 載體中,sgRNA 用于指導 Cas9 核酸酶在目標基因組區域誘導雙鏈斷裂,CBC 用于標記每個細胞,而 GBC 用于標記每個 sgRNA,UMI 用于標記每個轉錄本。Perutrb-seq 和 CRISP-seq 是第一個開發的 scCRISPR-seq 平臺。這些方法涉及 Perturb-seq 載體的復雜構建,包括復雜的克隆策略,有時還需要 gRNA 間隔區與其條形碼的解耦,這限制了它們的多功能性。CROPseq 優化了 Perturbseq 載體的設計,通過向 Perturb-seq 載體添加 Poly-A 尾部來檢測每個細胞中誘導的 sgRNA 與 mRNA 偶聯,這大大降低了 scCRISPR-seq 的復雜性和成本。然而,Hill 等人。(2018) 表明,由于這些研究的 Perturb-seq 載體設計,現有研究的慢病毒交換率僅為 50% 左右。因此,他們通過將向導 RNA 作為條形碼來優化 CROP-seq 載體設計,將交換率提高到 94%。由于 Perturb-seq 載體設計的限制,Perturb-seq 和 CRISP-seq 的每個慢病毒載體只能向細胞遞送單個編碼的 sgRNA,而 CROP-seq 能夠將成對的 sgRNA 遞送到細胞。也就是說,它們都與多個 sgRNA 的遞送不相容。為了解決這個問題,Replogle 等人。(2020) 設計了直接捕獲 Perturb-seq,其中表達的 sgRNA 與單細胞轉錄組一起測序,并能夠遞送多個 sgRNA。直接捕獲 Perturbseq 對于遺傳相互作用的機制解剖特別有價值。它進一步降低了 Perturb-seq 實驗的成本。Direct-seq 具有與直接捕獲 Perturb-seq 類似的功能,可實現 CRISPR 擾動及其轉錄讀數一起分析,并支持多個 sgRNA 的遞送。2022 年,Replogle 等人引入了基因組規模的 Perturb-seq 方法。(2022 年),能夠對影響 9,867 個基因的基因組規模遺傳擾動進行公正和全面的分析。這一突破促進了系統的基因功能分配和復雜細胞表型的探索。最近,利塔爾.(2023) 開發了 CRISPR-人類類器官-單細胞 RNA 測序 (CHOOSE) 系統。這種創新系統能夠進行遺傳破壞和單細胞轉錄組學,用于混合功能喪失篩選嵌合類器官。然而,上述所有 scCRISPR-seq 平臺都僅限于體外應用。相比之下,體內測定更具吸引力,因為它們與真實的有機條件更相似。因此開發了體內 Perturb-seq,這是 Perturb-seq 方案的一種變體,涉及在體內進行的合并擾動。此外,PoKI-seq (Roth et., 2020) 已經證明了體內研究重編程 T 細胞對實體瘤的免疫反應的可行性。最近,Santinha 等人。(2023) 開發了腺相關病毒 (AAV) 介導的直接體內單細胞 CRISPR 篩選,稱為 AAV-Perturb-seq,一種可用于體內遺傳擾動轉錄連鎖分析和表型分析的可調且廣泛適用的方法。
基于表觀基因組的 scCRISPR-seq
基于表觀基因組的 scCRISPR-seq 除了轉錄組應用外,還有基于表觀遺傳學的 scCRISPR-seq 平臺。2019 年開發了 Perturb-ATAC,這是一種將 CRISPR 干擾或敲除與單細胞染色質可及性分析相結合的方法,該方法基于通過測序檢測轉座酶可及性染色質 (ATAC-seq) 同時檢測 CRISPR 向導 RNA 和開放染色質位點。他們應用這種方法來確定各種反式調控因子的作用,包括 TFs、染色質修飾劑以及人類和病毒 ncRNA,這可能有助于剖析順式調控元件和 ncRNA 轉錄本已被證明對基因表達有影響的基因座(Cho 等人,2018b;Engreitz 等人,2016 年;Rubin 等人,2019 年)。Perturb-ATAC 將 scCRISPR-seq 研究擴展到表觀基因組領域,使 scCRISPR-seq 更強大、應用更廣泛。然而,Perturb-ATAC 受到高成本和低吞吐量的限制。為了應對這一限制,開發了 Spear-ATAC(Pierce 等人,2021 年)以實現顯著更高的細胞通量和大幅降低成本,提供了更實用的替代方案。此外,CRISPR-sciATAC (Liscovitch-Braueret al., 2021) 顯示出與 Spear-ATAC 相似的細胞通量和成本。然而,它對染色質可及性的細微變化表現出有限的敏感性。
多模態 scCRISPR-seq
多模式單細胞檢測提供了異質細胞群的高分辨率快照,但上述 scCRISPR-seq 平臺都僅限于一種模式,例如轉錄組或表觀基因組。因此,為了將該技術同時應用于多組學,開發了多模態 scCRISPR-seq。Xie et al. (2017) 通過索引 CRISPR 測序 (Mosaic-seq) 開發了Mosaic單細胞分析,以擾亂增強子并共同測量每個細胞的轉錄組及其誘導的 sgRNA。Mosaic-seq 提供了一種新穎的工具,以基于 inper擾動的方式詢問非編碼基因的功能。此外,Mimitou 等人。(2019) 通過測序開發了擴展的 CRISPR 兼容轉錄組和表位細胞索引 (ECCITE-seq),它允許同時檢測來自每個細胞的轉錄組、蛋白質、克隆型和 CRISPR 擾動。通過構建 ECCITEseq 抗體的 a49 標記面板來分析人外周血單核細胞 (PBMC),他們恢復了許多重要結果(Fanok等人,2018 年;Stoeckius 等人,2017 年),證明了 ECCITE-seq 結合免疫表型、克隆型和轉錄組信息的能力。空間轉錄組學能夠表征基因表達譜,同時保留有關空間組織背景的信息,這為生物學的不同領域提供了新的見解,例如神經科學、發育生物學和癌癥研究(Moses 和 Pachter,2022)(參見下面的空間部分)。最近,開發了一種新的多模態 scCRISPR-seq,稱為 Perturb-map (Dhainaut et., 2022),以通過成像和空間轉錄組學實現 CRISPR 篩選原位的多模態表型分析。Perturb-map 基于蛋白質條形碼 (Pro-Code) 系統,該系統使用幾個線性表位的三重體組合來創建更高階的唯一條形碼集(Wroblewska 等人,2018 年)。這些獨特的條形碼可以標記表達不同 CRISPR gRNA 的細胞。需要注意的是,Perturb-map 是唯一能夠實現體內 CRISPR 篩選與空間轉錄組相結合的 scCRISPR-seq 平臺,特別適用于鑒定腫瘤組成、組織和免疫的遺傳決定因素。Perturb-map 基于蛋白質條形碼 (Pro-Code) 系統,該系統使用幾個線性表位的三重體組合來創建更高階的唯一條形碼集(Wroblewska 等人,2018 年)。這些獨特的條形碼可以標記表達不同 CRISPR gRNA 的細胞。需要注意的是,Perturb-map 是唯一能夠實現體內 CRISPR 篩選與空間轉錄組相結合的 scCRISPR-seq 平臺,特別適用于鑒定腫瘤組成、組織和免疫的遺傳決定因素。Dhainaut 等人。(2022) 將 Perturb-map 應用于 TME 的研究。他們在肺癌小鼠模型中敲除 35 個基因,發現敲除 Tgfbr2 可以促進 TME 重塑和免疫排斥。
scCRISPR-seq 數據分析工具
scCRISPR-seq 數據包含豐富的擾動信息,這對于在單細胞水平上探索基因型和表型之間的關聯具有天然的優勢。例如,通過將 Perturb-seq 應用于 K562 細胞系,Adamson 等人。(2016) 表明,PERK 的擾動對未折疊蛋白質反應的影響比 ATF6 和 IRE1α 更大。Datlinger 等人 (2017) 在用 CROP-seq 激活 T 細胞受體 (TCR) 的條件下擾動了 Jurkat 細胞系中的 23 個轉錄因子,發現 LCK 、 ZAP70 和 LAT 的敲除對 TCR 激活信號傳導有很強的負面影響。然而,由于其固有的噪聲,scCRISPR-seq 數據的分析是一項重大挑戰。因此,已經開發了幾種生物信息學工具來幫助分析 scCRISPR-seq 數據(支持信息中的表 S12)。通常,這些 scCRISPR-seq 數據分析工具側重于三個部分(圖 16B):(i) 數據預處理,包括質量控制、歸一化和差異表達基因檢測,例如 MIMOSCA 、MUSIC和 SCREE 。(ii) 數據去噪,包括單細胞插補、轉義細胞過濾和復合因子分解,例如 MUSIC、mixscape 和 SCREE 。(iii) 功能分析,包括確定每個擾動影響的優先級,確定每個擾動的功能,推斷調節網絡和基因相互作用,例如 MUSIC、Normalisr、scMAGeCK、Pando和 GEARS。具體來說,LRICA是通過低秩矩陣分解來解碼數據的驅動信號/分量。MIMOSCA是一種計算sg RNA與每個基因之間關系的計算框架。LRICA 和 MIMOSCA 是作為原型開發的,沒有可執行且用戶友好的實現。因此,Duan 等人。(2019) 開發了 MUSIC,這是一個通用的計算框架,用于通過主題建模評估每個擾動的影響,它最初出現在機器學習和自然語言處理社區或特定文檔集中的潛在主題發現中。MUSIC 將基因型表型與對大量噪聲的耐受性聯系起來,并從三個角度分析 scCRISPR-seq 數據,即優先考慮基因擾動效應作為整體擾動效應、非功能性主題特異性方式,以及量化不同擾動之間的相關性。scMAGeCK 也是分析 scCRISPR-seq 數據的框架,它是從 MAGeCK 擴展而來的。scMAGeCK 包括兩個模塊,scMAGeCK-RRA 和 scMAGECK-LR,其中 scMAGeCK-RRA 用于通過負二項分布識別顯著富集的 sgRNA,scMAGeCK-LR 用于通過線性回歸評估受影響的基因。scMAGeCK 顯示出對假陽性的良好控制,并且比其他方法具有更好的靈敏度。除了 scCRISPR-seq 的通用計算框架外,一些工具還專注于數據去噪。例如,SCEPTRE 是為使用條件隨機化測試進行 scCRISPR-seq 數據校準而開發的。SCEPTRE 對 scCRISPR-seq 數據表現出良好的校準和靈敏度,產生了數百種由正交生物學證據支持的新調控關系。Mixscape 旨在通過混合判別分析過濾逃逸的細胞 (細胞誘導的 sgRNA,但不表現出擾動效應) 來提高 scCRISPR-seq 數據的信噪比。Normalisr 用于重建 scCRISPR-seq 數據的基因調控網絡。Wang 等人.(2022g) 強調了識別克隆細胞的重要性,因為它們可能導致 scCRISPR-seq 數據出現假陽性。SCREE 是 scCRISPR-seq 數據分析的綜合管道。與前面提到的最初專注于 scCRISPR-seq 中的數據去噪和挖掘的方法相比,GEARS 專門設計用于預測對單基因和多基因擾動的轉錄反應。這些方法大大增強了 scCRISPR-seq 數據的分析。
scCRISPR-seq 的應用
scCRISPR-seq 因其強大的能力而被廣泛應用于各個領域,包括連接基因型與表型、剖析遺傳調控以及研究腫瘤和自閉癥等非特異性疾病的遺傳機制。
連接基因型和表型
與傳統的 CRISPR 篩選只能識別具有非常不同表型的基因相比,scCRISPR-seq 具有揭示任何基因功能的能力。因此,scCRISPR-seq 天然適合大規模地將基因型與表型聯系起來。例如,Jaitin 等。(2016) 揭示了 22 個 TFs 對 CRISP-seq 刺激的脂多糖 (LPS) 刺激的出生骨髓細胞 (BMC) 的抗病毒、炎癥或發育過程的調節作用。Adamson 等人。(2016) 通過 Perturb-seq 系統分析了 K562 細胞中 83 個未折疊蛋白反應 (UPR) 相關基因的影響。此外,基因組規模的 Perturb-seq(Replogle等人,2022 年)提供了對遺傳擾動(9,867 個基因)的公正、全面的分析,有助于系統剖析與基因翻譯和核糖體生物發生相關的基因之間的關系。
剖析遺傳調控
scCRISPR-seq 還用于剖析基因組元件之間的復雜關系,包括編碼基因、轉錄因子、染色質調節因子、增強子和其他非編碼元件。例如,Adamson 等人。(2016) 使用 Perturb-seq 發現了三個 UPR 傳感器基因(ATF6、PERK 和 IRE1)之間的串擾。CROP-seq 擾亂了 LPS 刺激后 Jurkat 細胞中調節 TCR 激活的 TFs,并揭示了 TFs 之間的關系。此外,用于增強子擾動的 scCRISPR-seq,例如 Mosaic-seq (Xie et tal., 2017),可以發現新的增強子-基因對。此外,scCRISPR-seq 與 scATAC-seq 偶聯,如 Perturb-ATAC、Spear-ATAC 和 CRISPR-sciATAC 可以揭示人淋巴細胞和白血病細胞中的表觀遺傳景觀重塑劑
研究遺傳機制
有幾種體內 scCRISPR-seq 平臺可用,能夠研究腫瘤和自閉癥等非特異性疾病的遺傳機制。例如,Perturb-map (Dhainaut et., 2022) 有助于識別與腫瘤組成、組織和免疫相關的遺傳決定因素。Using Perturbmap, Dhainaut etal.(2022) 發現 TGFBR2 肺癌細胞的敲除促進了腫瘤微環境重塑和免疫排斥。Roth 等人。(2020) 使用 PoKI-seq 對增強 T 細胞抗腫瘤功能的嵌合抗原受體進行了篩選,提高了免疫抑制條件下的腫瘤浸潤和細胞殺傷率 內黑色素瘤。此外,Jin 等人。(2020b) 使用 invivo Perturb-seq 評估了 35 個與自閉癥譜系障礙/神經發育遲緩 (ASD/ND) 相關的 denovo 功能喪失風險基因。他們從神經元和神經膠質細胞類別中鑒定了細胞類型特異性和進化上保守的基因模塊。利塔爾。(2023) 還關注了這些高危自閉癥譜系障礙基因,他們揭示了它們對使用 CHOOSE 系統在嵌花類器官中決定細胞命運的影響。最近,Santinha 等人。(2023) 使用 AAV-Perturb-seq 系統分析了與成年小鼠大腦前額葉皮層中 22q11.2 缺失綜合征基因相關的表型景觀。他們確定了三個 22q11.2 連鎖基因,這些基因積極參與已建立的和以前未識別的體內神經元功能控制途徑。
總結
在本章中,我們對 scCRISPR-seq 進行了全面的回顧,分為三個不同的部分,其中包括 scCRISPR-seq 的類別、scCRISPR-seq 數據分析的工具以及 scCRISPR-seq 的顯著應用。scCRISPR-seq 一直是功能基因組學研究的強大方法 (Bock et al., 2022)。在本節中,我們根據其綜合組學方法將 scCRISPR-seq 分為三個主要類別:基于轉錄組的 scCRISPRseq、基于表觀基因組的 scCRISPR-seq 和多模態 scCRISPRseq。鑒于 scCRISPR-seq 數據中固有的噪聲,已經開發了多種生物信息學工具來幫助初始化分析,從而沒有顯著改進。
scCRISPR-seq 的多功能性使其在各個領域得到廣泛應用,提供了強大的功能,例如連接基因型與表型、剖析遺傳調控以及探索腫瘤和自閉癥等特定疾病的遺傳機制。然而,在生物學研究中更廣泛地采用之前,需要注意三個關鍵方面:(i) 降低復雜性和成本:應努力進一步簡化和降低 scCRISPR-seq 實驗的復雜性和成本。這將增強可擴展性和可及性,使更多實驗室能夠利用這項技術。(ii) 擴大對復雜組織和體內設置的適用性:雖然目前的 scCRISPR-seq 平臺主要針對細胞系,但迫切需要開發更強大的 scCRISPR-seq 平臺,這些平臺可以應用于更復雜的組織,包括類器官,以及理想的體內設置。這種擴展將使更廣泛的生物學研究成為可能。(iii) 降噪技術:隨著 scCRISPR-seq 平臺數量的增加,開發更強大的方法來破譯 scCRISPR-seq 數據中的固有噪聲變得至關重要。這些方法將有助于 scCRISPR-seq 結果的可靠性和可解釋性,進一步提高它們在不同研究環境中的實用性。
結尾
scRNA-seq 技術引起了世界各地許多科學家的廣泛關注,因為它具有在單細胞水平上研究細胞異質性的優勢。自新時代 inscRNA-seq 研究成立以來,已經過去了僅僅 14 年,在此之前,Tang 等人取得了初步的概念和技術突破。(2009) 在 2009 年。在測序技術和生物信息學不斷發展的推動下,scRNA-seq 研究領域目前正在經歷研究的激增。scRNA-seq 技術的成熟極大地促進了其他單細胞組學研究的進步。目前,單細胞組學檢測已擴展到基因組 (Dey et al., 2015)、表觀基因組 (Muto et al., 2021)、空間轉錄組學 (Chen et al., 2022)、蛋白質組學 (Petersonet al., 2017;Specht et al., 2021)、代謝組學 (Shrestha, 2020) 等多組學水平 (Angermueller et al., 2016),為單細胞水平研究提供更全面、更精細、更完整的分析策略。在這篇綜述中,我們總結了單細胞組學技術、數據分析及其應用的最新進展,概述了單細胞測序領域跨多個層次的前景。
在第 1 章中,我們全面概述了目前可用的 scRNA-seq 技術、實驗方法、數據分析程序及其在生物醫學領域的應用。最初,通過分離單細胞并獨立構建測序文庫來進行單細胞測序。這些單細胞測序技術只能檢測少量細胞(數到數百個),例如 Tang 方法、STRT-seq 和 SMART-seq(Islam 等人,2012 年;Ramsköld 等人,2012 年;Tang 等人,2009 年)。然而,隨著對測序技術的深入研究,基于條形碼標簽的單細胞鑒定已經出現,以及基于微滴或微孔的新型單細胞分離技術的出現,如 Drop-Seq 和 Cyto-Seq (Fan et., 2015a;Macosko 等人,2015 年),單細胞轉錄組測序已進入高通量時代。測序成本顯著降低,同時自動化和通量顯著提高。ScRNAseq 技術解決了細胞異質性問題,為臨床疾病尤其是腫瘤的個體化治療開辟了新途徑,促進了精準醫療的發展。然而,由于起始材料量少,scRNA-seq 存在 oflow 捕獲效率和高脫落率的局限性。與大量 RNA-seq 相比,scRNA-seq 產生的數據噪聲更大且可變性更強。盡管研究人員已經設計了多種工具來進行不同的 scRNA-seq 數據分析,但技術噪聲和生物變異(例如,隨機轉錄)仍然對 scRNA-seq 數據的計算分析構成巨大挑戰(Chen 等人,2019a)。因此,數據分析方法仍需進一步優化和改進。
與日益成熟的 scRNA-seq 技術相比,其他單細胞組學技術仍處于萌芽階段。在第 2、3、4 和 5 章中,我們重點介紹了過去十年中單細胞基因組、表觀基因組、蛋白質組學和代謝組學測序的最新工具、計算方法和應用。ScWGS 徹底改變了我們對遺傳變異及其對人類健康和疾病影響的理解。它的快速發展加速了基因組研究,實現了個性化醫療,并為疾病的遺傳基礎和人類基因組多樣性提供了有價值的見解。細胞在染色質可及性、核小體定位、組蛋白修飾和 DNA 甲基化方面表現出廣泛的異質性。在單細胞樣品中繪制這些表觀基因組信息對于發育生物學、癌癥研究以及很快的發展非常重要。單細胞表觀基因組測序方法的進步使單細胞染色質狀態的高分辨率圖譜成為可能。然而,如今,單細胞表觀基因組技術存在數據丟失的問題。因此,盡管單個細胞表觀基因組數據集是聚類分析和基于大量目標位點集合揭示細胞異質性的強大資源,但它們提供單個目標位點信息的能力非常有限(Carter 和 Zhao,2021)。因此,在未來的研究中,需要提高各種單個細胞表觀基因組測定中染色質靶位點的覆蓋率,這將有助于理解整個細胞水平和單個特異性位點的細胞異質性。單細胞蛋白質組學由于其成分復雜、豐度低、動態范圍寬、缺乏擴增能力,處于爆發性發展的早期階段。就在 2019 年,對單細胞蛋白質組的分析被描述為“夢想”,但今天已經開發了幾種有前途的工具(Marx,2019)。我們相信,隨著可及性的優化和通量的進一步提高,單細胞蛋白質組學在科學和臨床研究中真正的大規模應用,如器官圖譜、藥物篩選和精確的疾病分類,是觸手可及的。單細胞代謝組學用于鑒定單細胞中代謝物的組成,測量其豐度,并研究其動態變化。同時,代謝組代表基因組、轉錄組和蛋白質組的下游產物,并提供了功能的更直接和動態快照(Shrestha,2020 年)。總體而言,單細胞組學技術仍處于萌芽階段,它們將繼續蓬勃發展。
單個細胞是生命的基本單位。對單個細胞進行多組學分析可以深入了解細胞的表型、疾病狀態和環境影響。在第 6 章、第 7 章和第 8 章中,我們全面總結了多組學的綜合分析、scRNA-seq 和 CRISPR 篩選的聯合應用以及空間轉錄組。在復雜的生物過程中,例如腫瘤發生和衰老,異質性發生在不同的層面,包括基因組、轉錄組、蛋白質組和表觀基因組。如果僅從單個細胞 atatime 分析一個成分,則只能檢測到基因調控網絡的局部概況,而無法準確預測復雜的全球情況。在這種情況下,多組學技術凸顯其獨特的優勢,可以在復雜組織的研究中提供更完整的基因調控網絡圖譜。對于空間轉錄組學,它能夠在保留空間信息的情況下測量基因表達,這將有利于研究細胞間關系和發現空間背景下的新調控機制。此外,空間轉錄組學使探索細胞命運決定的空間調控機制和組織模式的結構成為可能。與傳統的 CRISPR 雜交篩選相比,scRNA-seq 和 CRISPR 的組合不僅可以在單個實驗中篩選數千個 gRNA,還可以同時捕獲擾動的全轉錄組數據,以最清楚地了解細胞類型特異性基因功能和通路分析。因此,這些技術的結合可以更好、更深入地理解關鍵的生物過程和機制,這是未來單細胞技術發展的重要方向。
如今,單細胞組學技術在通量和分辨率方面都取得了重大進步。展望未來,單細胞技術發展的主要趨勢是提高單細胞分選的效率和通量,增強測序覆蓋率和靈敏度,實現高通量多組學研究,并開發更多自動化的單細胞技術平臺,這將有助于降低單細胞技術的成本和技術門檻。單細胞技術有望在科學研究和研究轉化領域得到廣泛應用,并將對健康監測、疾病診斷和治療做出巨大貢獻。
參考文獻:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0