子宮內膜異位癥(EM)是育齡女性常見疾病,在不孕女性中發病率為20%-50%,EM患者不孕風險是無EM者的4 倍,但EM相關不孕機制尚未完全闡明,目前認為卵泡發育異常和卵母細胞質量下降是主要誘因。氧化應激是導致EM患者卵泡發育異常和卵母細胞質量降低的重要因素,可通過抑制線粒體功能、干擾脂質代謝、加速顆粒細胞(GCs)衰老等損害卵母細胞數量和質量;GCs作為卵泡重要組成,通過縫隙連接和旁分泌為卵母細胞提供營養和能量,其衰老與EM相關不孕密切相關。
2025年8月,浙江大學醫學院附屬邵逸夫醫院張松英、戴永東、周楓共同通訊在Advanced Science (IF 14.1)在線發表題為“EETs Reduction Contributes to Granulosa Cell Senescence and Endometriosis-Associated Infertility via the PI3K/AKT/mTOR Signaling Pathway”的研究論文。首次系統闡明EM相關不孕中的氧化脂質譜,明確14,15-EET的抗GC衰老作用,揭示 ROS-EZH2/H3K27Me3-EPHX2-EET惡性循環機制,為EM相關不孕提供新治療靶點,并證實了TPPU和BEZ-235的潛在治療價值。

· 維真助力·
基因信息
EZH2:組蛋白甲基轉移酶
EPHX2:可溶性環氧化物水解酶
病毒產品
LV-sh-EZH2、LV-sh-EPHX2、LV-sh-NC ;
LV-EZH2、LV-EPHX2、LV-NC
感染細胞
小鼠卵巢顆粒細胞(mGC)
MOI
10
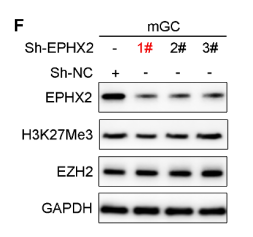
WB檢測LV-sh-EZH2、LV-sh-EPHX2的敲低效果
研究結果
1、Ezh2活性降低通過減少Ephx2基因啟動子的H3K27Me3修飾上調EPHX2表達水平
研究人員通過對不孕婦女的卵泡液(FF)、腹膜液 (PF) 和卵巢顆粒細胞 (GC) 進行LC-MS/MS分析,發現氧化脂質14,15-EET水平在EM患者中顯著下降,并與IVF結局呈正相關。使用14,15 EET預處理可通過提高mGCs的抗氧化能力和ATP水平,減輕過度氧化應激引起的線粒體功能障礙和ROS誘導的卵巢GC衰老。EETs由花生四烯酸(AA)經細胞色素P450單加氧酶(如CYP3A5)合成,再通過可溶性環氧化物水解酶(EPHX2)代謝為低活性的DHETs。研究發現,EM-GCs與Con-GCs中,CYP3A5的mRNA表達水平無顯著差異;但EPHX2的mRNA和蛋白表達水平均顯著升高,提示EM患者卵泡液中EETs減少源于EPHX2介導的代謝增加,而非合成異常。EM-GCs中,組蛋白甲基轉移酶EZH2的蛋白表達水平顯著低于Con-GCs,同時其催化產生的抑制性組蛋白修飾標志物H3K27Me3水平也顯著下降;而細胞衰老標志物蛋白P21的表達水平顯著升高,暗示EZH2/H3K27Me3下調與EM-GCs衰老及EPHX2上調存在關聯。通過慢病毒介導的Sh-EZH2敲低mGCs中EZH2表達后,qRT-PCR結果顯示Ezh2 mRNA水平顯著降低,而Ephx2 mRNA水平顯著升高;Western blot同樣證實Sh-EZH2處理組的EZH2蛋白水平降低,EPHX2蛋白水平顯著升高。ChIP-PCR進一步驗證H3K27Me3與EPHX2啟動子的結合,表明EM-GC中Ezh 2活性下降導致Ephx2啟動子區域基于H3K27Me3的組蛋白甲基化減少,從而導致EPHX2蛋白表達水平增加,促進14,15-EET代謝,最終降低EM卵泡中14,15-EET水平。
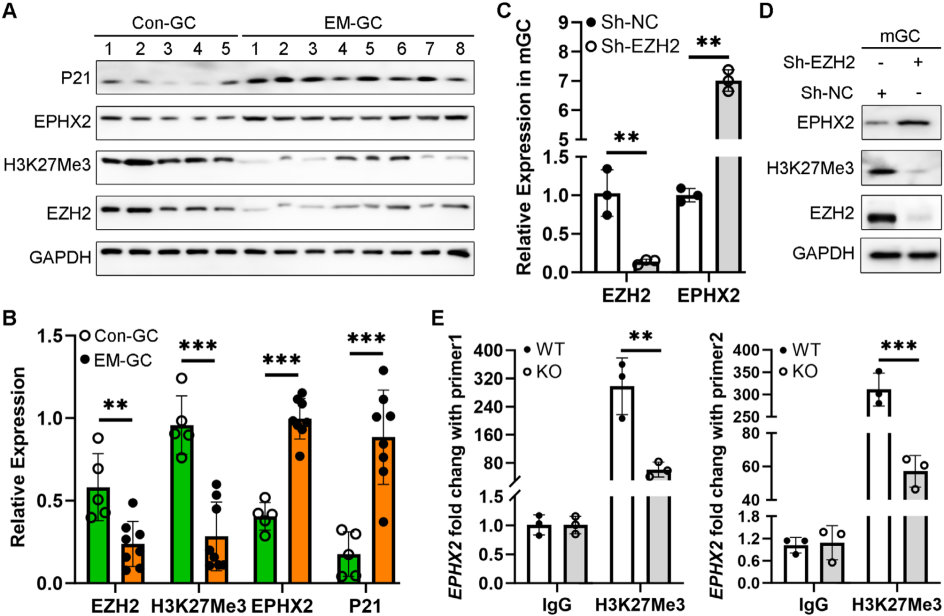
EZH2/H3K27Me3組蛋白甲基化降低導致EM-GCs中EPHX2表達水平升高
2、EPHX2過表達加劇卵巢GC衰老,而其抑制劑TPPU減輕ROS誘導的細胞衰老
在誘導氧化應激后過表達EZH2,導致EPHX2和衰老標記蛋白的表達水平降低。有趣的是,在氧化應激條件下,EPHX2敲低上調EZH2/H3K27Me3表達水平,但抑制衰老標記蛋白表達水平。此外,EPHX2敲低顯著升高14,15-EET濃度,并逆轉ROS誘導的GC衰老表型。進一步在體外過表達EPHX2會加劇GCs衰老,而其特異性抑制劑TPPU可通過上調EETs水平,緩解ROS誘導的GCs衰老。接下來,研究團隊建立EM小鼠模型,探索了TPPU處理(驗證了無明顯毒性)是否可以影響體內細胞衰老,結果顯示TPPU可上調EM小鼠GCs中EETs、EZH2及H3K27Me3水平,降低EPHX2及衰老標記蛋白的表達水平。這些數據表明TPPU在減輕體內mGC衰老中的重要作用,該功能可能通過上調EET水平來實現。進一步研究證實TPPU處理可延緩EM小鼠生育能力下降,通過調控卵巢微環境,顯著改善卵丘-卵母細胞復合體(COC)的擴張異常,并抑制卵母細胞中活性氧的積累。
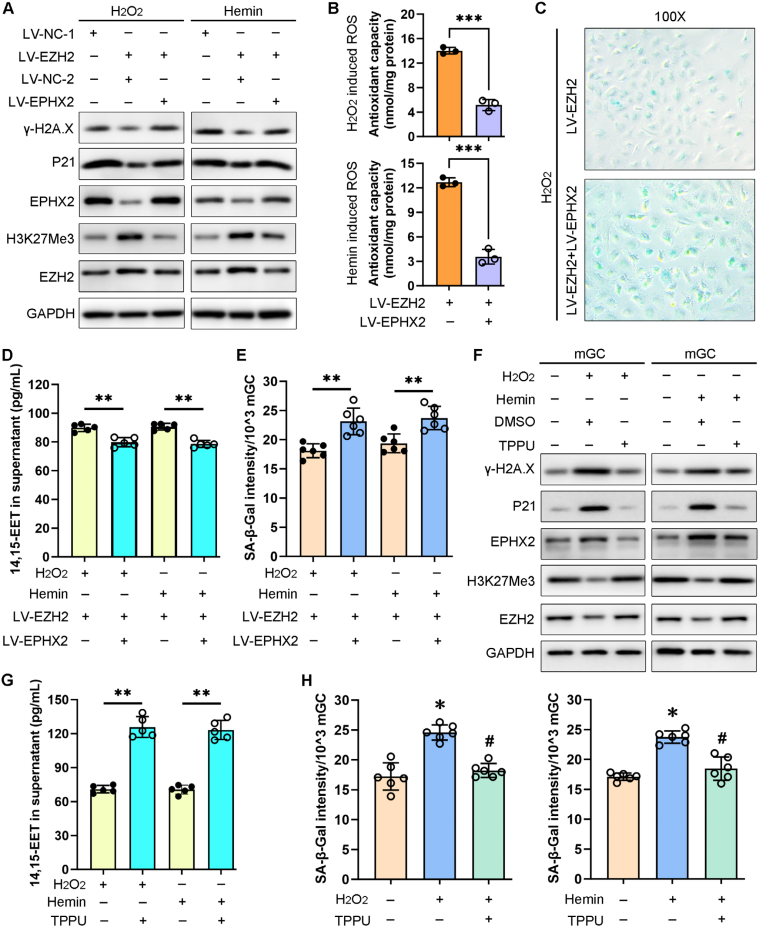
EPHX2過表達加重mGC衰老,而TPPU在體外挽救ROS誘導的細胞衰老
3、14,15-EET通過抑制EM中PI3K/AKT/mTOR信號通路的過度激活來減輕GC衰老
對EM-mGCs和Con-mGCs的卵巢GCs進行RNA測序,基因集富集分析顯示,與 Con-mGCs相比,EM-mGCs中衰老和細胞衰老相關基因集顯著富集,同時PI3K-Akt信號通路和mTOR信號通路被顯著激活。為明確PI3K/AKT/mTOR通路在14,15-EET抗衰中的核心作用,在14,15-EET預處理并誘導氧化應激的mGCs中,使用AKT特異性激活劑 SC79重新激活 AKT。結果顯示,SC79 處理后,衰老標志物蛋白表達水平顯著回升,細胞衰老表型再次出現,證實14,15-EET對GC衰老的緩解作用依賴于對PI3K/AKT/mTOR通路的抑制。進一步用14,15-EET處理EM來源的mGC后,RNA-seq和KEGG分析顯示,PI3K-AKT信號通路的富集程度顯著降低。Western blot驗證,在氧化應激條件下,14,15-EET預處理能有效抑制PI3K p85、p-AKT和p-mTOR的過度激活。這些結果共同表明PI3 K/AKT/mTOR信號通路在EM相關不育中的重要作用,這意味著14,15-EET可能通過抑制EM-GC中該通路的過度激活而起作用。在EM小鼠模型及體外實驗中,雙靶點I類PI3K/mTOR抑制劑BEZ-235通過抑制PI3K/AKT/mTOR信號通路過度激活,顯著緩解GCs衰老,并改善COC擴張異常。
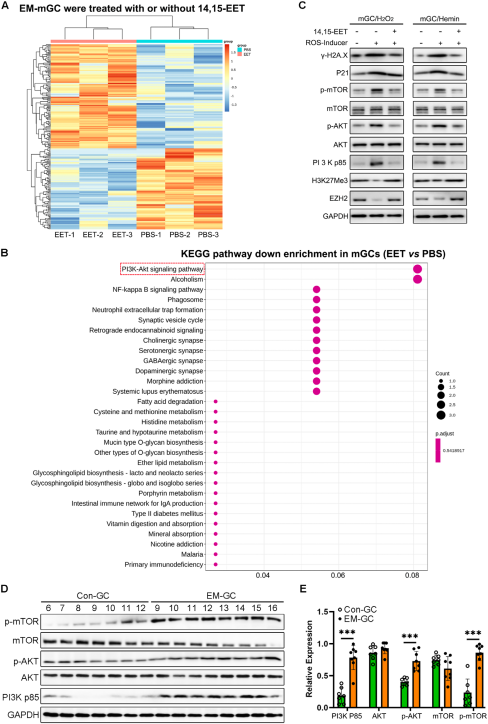
14,15-EET通過抑制PI3K-AKT-mTOR信號通路過度激活挽救ROS誘導的mGC衰老
研究結論
本研究首次闡明ROS-EZH2/H3K27Me3-EPHX2-EET這一反饋環路形成了一個惡性循環,該循環可促進卵巢顆粒細胞衰老,并參與子宮內膜異位癥相關不孕的發生發展。14,15-EET可通過抑制EM-GCs中PI3K-AKT-mTOR信號通路的異常激活,緩解顆粒細胞衰老并改善生育能力。